
The rare diseases that
need our attention
We focus on rare diseases where the burden is significant, and the need for better treatment options is clear. By combining scientific insight with a deep understanding of patient experience, we work to advance therapies that can make a meaningful difference in patients’ lives.
Explore our therapeutic areas of focus below.
Rare liver diseases are often complex, progressive, and deeply disruptive to daily life. We have built a leading portfolio in cholestatic liver diseases—addressing a broad range of conditions—while expanding into other rare liver diseases where unmet need remains high. Through our expertise, we develop therapies that target both the underlying disease and the symptoms that matter most to patients.
Select a condition to learn more.
Alagille Syndrome (ALGS)
ALGS is a rare, multisystem disease caused by abnormalities in bile ducts that can lead to progressive liver disease.1
Bile ducts carry bile from the liver to the gallbladder and small intestine.1 Bile has many purposes, including aiding in the digestion of fats and helping with the absorption of fat and certain types of vitamins (A, D, E, and K).2 In people with ALGS, the bile ducts may be too narrow, too few, or missing altogether. This can result in cholestasis: a condition where the flow of bile from the liver is slowed or blocked, causing bile acids to build up in the body.3,4
Over time, the buildup of bile acids in the liver can lead to long-term problems, including inflammation, scar tissue, and injury, as well as the risk for transplant.3,5-7
Approximately 1 in every 30,000 children in the United States and Europe are born with ALGS.8
Signs of ALGS typically begin during infancy and symptoms attributed to cholestasis include2:
- Yellow skin or eyes (jaundice)
- Cholestatic pruritus (itch)
- Stunted growth
Up to 88% of people with ALGS experience cholestatic pruritus (itch).9 Cholestatic pruritus is often a very challenging symptom for people with ALGS, beyond the scratching. It can present in patients differently and range in severity. Signs and symptoms related to the itch can look different during the day and/or night, vary among patients, or even change with age.9,10
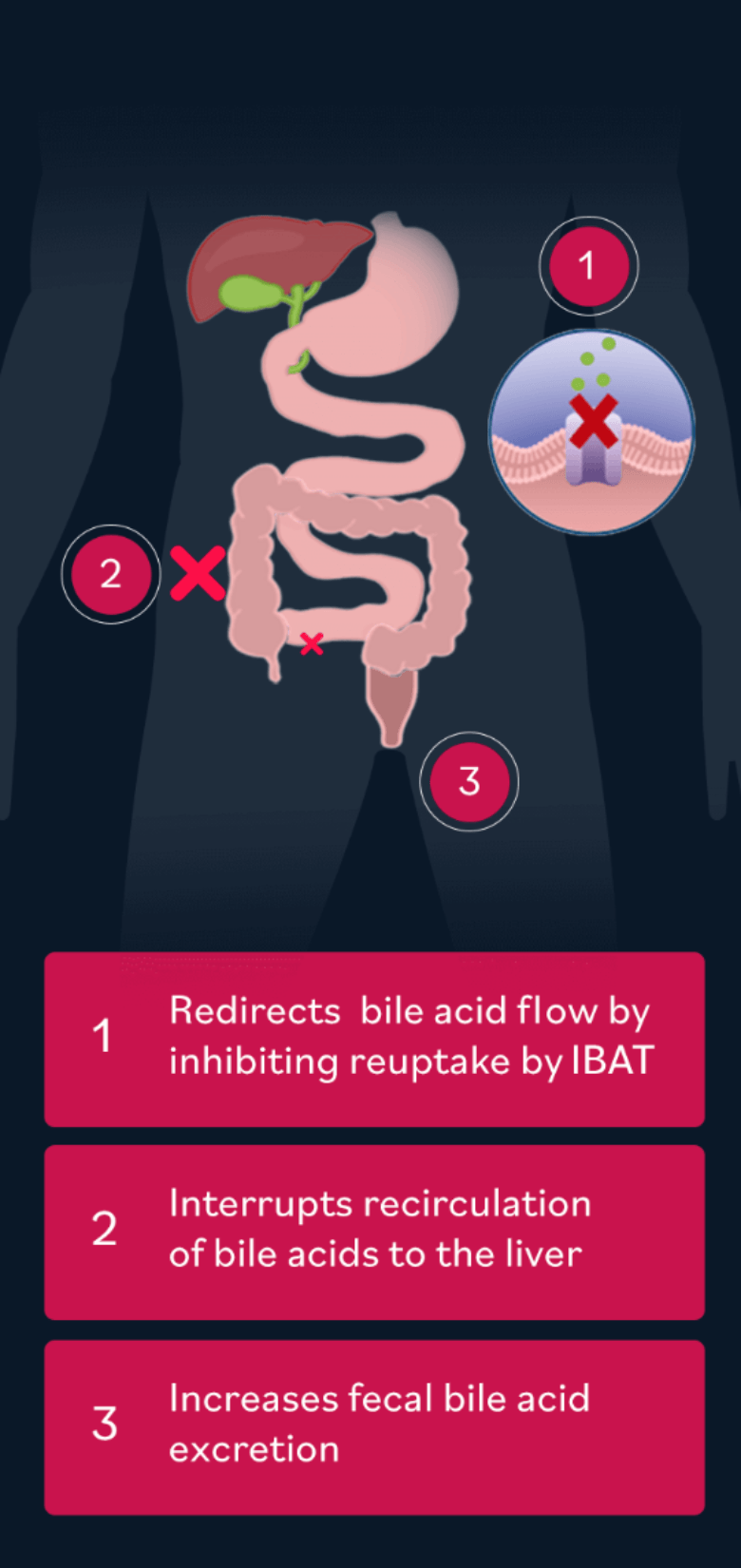
Ileal bile acid transporter (IBAT) inhibition has been approved to treat cholestatic pruritus in patients with ALGS. IBAT inhibition seeks to help redirect bile acids to excrete more in the feces, leading to lower circulating levels and reduced bile buildup in the liver.
References
- Definition & facts for Alagille syndrome. National Institute of Diabetes and Digestive and Kidney Diseases. Updated June 2018. Accessed June 4, 2021. https://www.niddk.nih.gov/health-information/liver-disease/alagille-syndrome/definition-facts
- Alagille syndrome. Johns Hopkins Medicine. Accessed April 29, 2021. https://www.hopkinsmedicine.org/health/conditions-and-diseases/alagille-syndrome
- Kamath BM, Stein P, Houwen RHJ, Verkade HJ. Potential of ileal bile acid transporter inhibition as a therapeutic target in Alagille syndrome and progressive familial intrahepatic cholestasis. Liver Int. 2020;40(8):1812-1822. doi:10.1111/liv.14553
- Alagille syndrome. MedlinePlus. Updated December 1, 2023. Accessed February 7, 2024. https://medlineplus.gov/genetics/condition/alagille-syndrome
- Cai SY. The role of bile acids in cholestatic liver injury. Ann Transl Med. 2021;9:737. doi:10.21037/atm-20-5110
- Karpen SJ, Kelly D, Mack C, Stein P. Ileal bile acid transporter inhibition as an anticholestatic therapeutic target in biliary atresia and other cholestatic disorders. Hepatol Int. 2020;14(5):677-689. doi:10.1007/s12072-020-10070-w
- Li T, Chiang J. Bile acid-induced liver injury in cholestasis. In: Ding WX, Yin XM, eds. Cellular Injury in Liver Diseases. Cell Death in Biology and Diseases. Springer; 2017:143-172. doi:10.1007/978-3-319-53774-0_7
- Vandriel SM, Li LT, She H, Wang JS, et al; Global Alagille Alliance (GALA) Study Group. Natural history of liver disease in a large international cohort of children with Alagille syndrome: results from the GALA study. Hepatology. 2023;77(2):512-529. doi:10.1002/hep.32761
- Kamath BM, Baker A, Houwen R, Todorova L, Kerkar N. Systematic review: the epidemiology, natural history, and burden of Alagille syndrome. J Pediatr Gastroenterol Nutr. 2018;67(2):148-156. doi:10.1097/MPG.0000000000001958
- Langedijk JAGM, Beuers UH, Oude Elferink RPJ. Cholestasis-associated pruritus and its pruritogens. Front Med (Lausanne). 2021;8:639674. doi:10.3389/fmed.2021.639674
Progressive familial intrahepatic cholestasis (PFIC)
PFIC is a group of rare, inherited, genetic diseases that mostly affect the liver. PFIC is characterized by impaired bile acid secretion or transport, leading to progressive cholestatic liver disease. Over time, the buildup of bile acids in the liver can lead to long-term problems, including inflammation, scar tissue, and injury, as well as the risk for transplant.1
PFIC is classified into three main subtypes along with several rare subtypes. These subtypes are defined by underlying genetic mutations, such as in the ABCB11 or ABCB4 genes, which, when altered, cause bile acids to accumulate in the liver.2,3 Across all PFIC subtypes, there is an inhibition of bile flow between the liver and small intestine, resulting in a persistent state of cholestasis.4,5
Signs of PFIC can begin as early as infancy and can result in:
- Cholestatic pruritus (itch)2
- Stunted growth1,2
- Vitamin deficiency1
- Yellow skin or eyes (jaundice)2
- Progressive liver damage2
- Liver failure if left untreated2
Though PFIC typically appears in infancy or early childhood, with some types, like PFIC3, symptoms may not present until adulthood. PFIC can also be missed in adults, and symptoms like cholestatic pruritus are often normalized or overlooked, adding to diagnosis challenges.2,6
Up to 100% of people with PFIC are affected by cholestatic pruritus and it is also the most burdensome symptom.7 Uncontrolled cholestatic pruritus causes more than just skin damage. Patients can also struggle with fatigue, irritability, physical discomfort, decreased physical function, impaired school performance, and negative impact on social activities.1,7
There are fewer than 1,000 pediatric patients with PFIC in either the United States or Europe.8
Ileal bile acid transporter (IBAT) inhibition has been approved to treat cholestatic pruritus in patients with PFIC.* IBAT inhibition seeks to help redirect bile acids to excrete more in the feces, leading to lower circulating levels and reduced bile buildup in the liver.
*IBAT is not recommended in a subgroup of PFIC patients with non-functional or complete absence of BSEP protein.
References
- Srivastava A. Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol. 2014;4(1):25-36. doi:10.1016/j.jceh.2013.10.005
- Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009;4:1. doi:10.1186/1750-1172-4-1
- Vinayagamoorthy V, Srivastava A, Sarma MS. Newer variants of progressive familial intrahepatic cholestasis. World J Hepatol. 2021;13(12):2024-2038. doi:10.4254/wjh.v13.i12.2024
- Felzen A, Verkade HJ. The spectrum of progressive familial intrahepatic cholestasis diseases: update on pathophysiology and emerging treatments. Eur J Med Genet. 2021;64(11):104317. doi:10.1016/j.ejmg.2021.104317
- Kamath BM, Stein P, Houwen RHJ, Verkade HJ. Potential of ileal bile acid transporter inhibition as a therapeutic target in Alagille syndrome and progressive familial intrahepatic cholestasis. Liver Int. 2020;40(8):1812-1822. doi:10.1111/liv.14553
- PFIC diagnosis – types of tests. PFIC Network. Accessed March 30, 2026. https://www.pfic.org/learn-about-pfic-disease/pfic-diagnosis/
- Baker A, Kerkar N, Todorova L, Kamath BM, Houwen RHJ. Systematic review of progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2019;43(1):20-36. doi:10.1016/j.clinre.2018.07.010c
- Data on File. Mirum Pharmaceuticals.
Primary sclerosing cholangitis (PSC)
PSC is a rare cholestatic liver disease characterized by inflammation and scarring of the bile ducts, which lead to hardening and narrowing (strictures) in the bile ducts inside or outside the liver. This prevents the bile from flowing properly, ultimately requiring a liver transplant for many patients.1 These blockages can cause bile acids to build up in the body—a condition known as cholestasis. This leads to higher levels of bile acids in the blood, which are linked to common symptoms like itching and fatigue.2
PSC affects approximately 10 in 100,000 people, and nearly 70% of those patients also have inflammatory bowel disease.3,4
Signs and symptoms of PSC change as the disease progresses and may include:
EARLY SYMPTOMS
- Itch (pruritus)5
- Fatigue5
- Brain fog6
- Upper right quadrant abdominal pain5
- Episodes of cholangitis (an infection within the liver)7
- Chills and fever5
LATER SYMPTOMS
- Ascites (swollen abdomen with fluid)8
- Enlarged spleen (splenomegaly)4
- Yellowing of skin and eyes (jaundice)8
- Unintended weight loss4
- Liver failure5
Up to 91% of PSC patients experience itch at some point during the course of their disease, often linked to sleep disturbance, fatigue, and mood changes.9 Many PSC patients will experience pruritus (itching) and it tends to worsen as the disease progresses. While itch is a common symptom of PSC, it is often underreported.10,11
Despite the impact of PSC on a patient’s quality of life, there are currently no approved treatments that can slow or stop PSC from progressing or treat the persistent itch and fatigue that typically accompany the disease.8 To address this unmet need, we are investigating a selective inhibitor of the ileal bile acid transporter (IBAT) to treat cholestasis. IBAT inhibition seeks to help redirect bile acids to excrete more in the feces, leading to lower circulating levels and reduced bile buildup in the liver.
References
- Mayo Foundation for Medical Education and Research. Primary sclerosing cholangitis: Symptoms and causes. Mayo Clinic. Published June 21, 2023. Accessed June 2, 2025. https://www.mayoclinic.org/diseases-conditions/primary-sclerosing-cholangitis/symptoms-causes/syc-20355797
- Sanjel B, Shim WS. Recent advances in understanding the molecular mechanisms of cholestatic pruritus: a review. Biochim Biophys Acta Mol Basis Dis. 2020;1866(12):165958. doi:10.1016/j.bbadis.2020.165958
- Sohal A, Kayani S, Kowdley KV. Primary sclerosing cholangitis: epidemiology, diagnosis, and presentation. Clin Liver Dis. 2024;28(1):129-141. doi:10.1016/j.cld.2023.07.005
- Primary sclerosing cholangitis: MedlinePlus Genetics. Accessed April 1, 2026. https://medlineplus.gov/genetics/condition/primary-sclerosing-cholangitis/
- What is primary sclerosing cholangitis? Cleveland Clinic. Accessed March 31, 2026. https://my.clevelandclinic.org/health/diseases/23569-primary-sclerosing-cholangitis
- Arndtz K, Cameron M, Hirschfield G, Parry J, Greenfield S. What are the lived healthcare experiences of patients with primary sclerosing cholangitis? A community-based qualitative interview study. BMJ Open. 2025;15(2):e082498. doi:10.1136/bmjopen-2023-082498
- Chazouilleres O, Beuers U, Bergquist A, et al. EASL Clinical Practice Guidelines on sclerosing cholangitis. J Hepatol. 2022;77(3):761-806. doi:10.1016/j.jhep.2022.05.011
- Lindor KD, Kowdley KV, Harrison ME; American College of Gastroenterology. ACG clinical guideline: primary sclerosing cholangitis. Am J Gastroenterol. 2015;110(5):646-659. doi:10.1038/ ajg.2015.112
- Kowdley KV, Safer R, Gomel R, et al. Impact of pruritus in primary sclerosing cholangitis (PSC): a multinational survey. Poster presented at: The International Liver Congress; 2022. Accessed June 2, 2025 https://www.postersessiononline.eu/173580348_eu/congresos/ILC2022/aula/-THU_442_ILC2022.pdf
- Dean R, Yazdanfar M, Zepeda J, et al. Treatment of pruritus in primary sclerosing cholangitis: analysis of the consortium for autoimmune liver disease registry. Hepatol Commun. 2025;9(5):e0703. doi:10.1097/HC9.0000000000000703
- Hussain N, Motta RV, Gungabissoon U, et al. Pruritus is common in primary sclerosing cholangitis, persists over time, and its intensity is associated with disease severity: a multicentre, prospective observational study. Hepatol. Published online December 5, 2025.
Primary biliary cholangitis (PBC)
PBC is a rare cholestatic liver disease that causes the small bile ducts in the liver to become injured and inflamed. When bile ducts become injured, bile builds up (cholestasis) and potentially causes progressive liver damage, with the need for liver transplant in some patients.1 Cholestasis is linked to common symptoms such as itch and fatigue.2
PBC is thought to be an autoimmune disease in which the body attacks its own bile ducts. People with a family member who has PBC may also have a higher chance of developing the disease.1
Early and general symptoms include3:
- Fatigue
- Itch (pruritus)
- Abdominal pain
Later signs and symptoms of PBC include3:
- Jaundice
- Enlarged spleen (splenomegaly)
- Swelling in the feet and ankles
People living with PBC typically rate the itch and fatigue they experience as moderate to severe.4,5 The burdensome itch can significantly impact a patient’s quality of life, affecting their social and emotional well-being and contributing to sleep disruption that worsens fatigue.5 While itch and fatigue are common symptoms of PBC, they are often underreported.4,5
There is no cure for PBC, but medications may help manage the disease and slow the progression of liver damage. Other medications are typically prescribed to help manage a person’s symptoms, including itch. When medications do not help, liver transplantation may be necessary.6 Mirum is investigating a selective inhibitor of the ileal bile acid transporter (IBAT) to treat cholestasis. IBAT inhibition seeks to help redirect bile acids to excrete more in the feces, leading to lower circulating levels and reduced bile buildup in the liver.
References
- Mayo Foundation for Medical Education and Research. Primary biliary cholangitis. Mayo Clinic. Published November 14, 2023. Accessed June 2, 2025. https://www.mayoclinic.org/diseases-conditions/primary-biliary-cholangitis/symptoms-causes/syc-20376874
- Faisal A. Understanding fatigue and pruritus in primary biliary cholangitis. Clin Liver Dis (Hoboken). 2024;23(1):e0216. doi:10.1097/CLD.0000000000000216
- Lindor KD, Bowlus CL, Boyer J, Levy C, Mayo M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology. 2019;69(1):394-419. doi:10.1002/hep.30145
- Al-Harthy N, Kumagi T, Coltescu C, Hirschfield GM. The specificity of fatigue in primary biliary cirrhosis: evaluation of a large clinic practice. Hepatology. 2010;52(2):562-570. doi:10.1002/hep.23683
- Gungabissoon U, Smith HT, von Maltzahn R, et al. Pruritus in primary biliary cholangitis is under-recorded in patient medical records. BMJ Open Gastroenterol. 2024;11(1):e001287. doi:10.1136/bmjgast-2023-001287
- National Organization for Rare Disorders. Primary biliary cholangitis. NORD. Updated April 6, 2020. Accessed June 1, 2025. https://rarediseases.org/rare-diseases/primary-biliary-cholangitis/
Rare genetic diseases are often lifelong, affecting multiple systems and placing a significant burden on patients and families. We are focused on conditions driven by well-defined genetic and metabolic pathways, advancing treatments that target underlying disease biology with the goal of delivering meaningful impact for communities that have long been overlooked and underserved.
Select a condition to learn more.
A pipeline builtfor impact
Our therapeutic focus areas are reflected in a pipeline designed with intention—advancing programs where scientific insight, patient need, and clinical clarity align.
From approved medicines to late-stage development, we are building a portfolio focused on delivering meaningful progress for patients living with rare diseases.
